
For Physicians
WARNING:
IMAGES AND TOPICS COVERED ON THIS PAGE MAY BE CONSIDERED GRAPHIC TO SOME AUDIENCES, AND ARE INTENDED FOR PHYSICIANS ONLY. VIEWER DISCRETION IS ADVISED.
This page is mainly intended to help physicians inform their patients about Degos disease.
That being said, medical terms are explained in brackets so that patients who wish to learn these concepts on their own, can do so in order (for example) to understand doctors’ reports and scientific literature.
Overview
Degos disease, also known as Malignant Atrophic Papulosis (MAP), Atrophic Papulosis, or Kohlmeier-Degos disease, is an ultra-rare disease which can exist in a benign, cutaneous (skin only) or systemic, potentially malignant form. It was initially described by Köhlmeier in 1941 and documented as a distinct illness by Degos in the same year.
Symptoms normally appear between ages 20-50, but can affect any age group. The illness predominantly occurs sporadically but on rare occasions has affected several members of the same family.
The systemic form of Degos disease affects one or more body organs, most commonly the gastrointestinal tract or brain. Complications can include bowel perforation and various neurologic and visual symptoms.
Diagnosing Degos disease
Skin lesions precede internal involvement and begin as small erythematous (red) macules, commonly affecting the trunk and upper extremities. Over days, the centers of the lesions become atrophic (sunken), with an older lesion therefore displaying the classic porcelain-white center with an erythematous rim. Characteristically, palmar (palms of hands), plantar (bottoms of feet), and facial regions remain unaffected.
The characteristic skin changes make clinical diagnosis possible, but a skin biopsy is required for conclusive pathologic diagnosis. Laboratory tests are not necessary for diagnosis, as coagulating disturbances may be present.
Our Goal: Promote early recognition/diagnosis for better patient outcomes.
The sooner skin lesions are noticed, the sooner next steps and treatments can begin, if necessary. If the disease remains confined to the skin, it is benign. If systemic disease develops, acute complications and even death may occur.
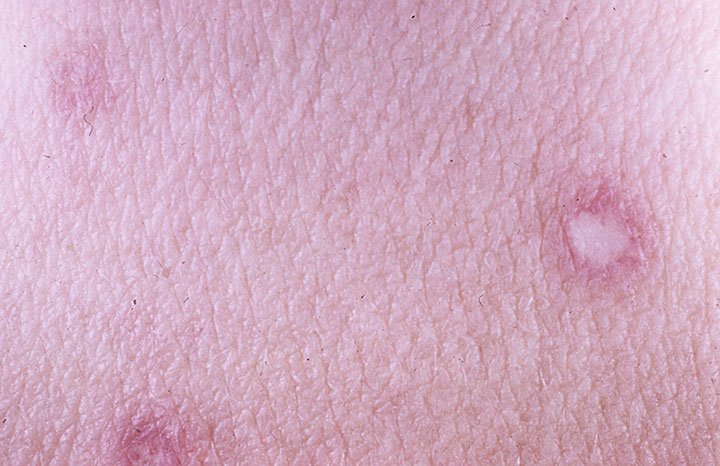




Degos disease cutaneous skin lesions.

Early diagnosis can make a big difference.
Cutaneous Path to Diagnosis
-

STEP 1
If a lesion appears on your patient’s skin, perform a skin biopsy.
-

STEP 2
Send the biopsy slides to Doctor Cynthia Magro for analysis.
Doctor Magro is the world expert on disease pathology.
Contact her, especially if there is any uncertainty to diagnosis.
-

STEP 3
If cutaneous Degos disease is confirmed by Doctor Magro, help enroll your patient in the research protocol at the NIH (National Institutes of Health).
Help your patient pursue referrals to NIH:
Download the NIH Referral Process Q&A PDF HereIf the patient is too young to be enrolled, the work-up can be done by a specialist (dermatologist or pediatrician).
-

STEP 4
If your patient is experiencing abdominal pain, SEE PATH BELOW AND TAKE IMMEDIATE ACTION IN TREATMENT.
If patient presents with unexplained abdominal pain…
Gastrointestinal Path to Diagnosis
-
STEP 1
TAKE IMMEDIATE ACTION
If your patient is experiencing abdominal pain, a URGENT laparoscopy should be performed to rule out systemic involvement.
If lesions are detected, they need medication RIGHT AWAY.
The surgeon performing the laparoscopy should familiarize themselves with the appearance of the bowel lesions here, before the procedure.
Also review the article and watch the Laparoscopy training video below.
-

STEP 2
ASSESS THE SITUATION
If your patient has an “acute abdomen” they need surgical exploration.
If multiple lesions are observed, they are at IMMINENT HIGH RISK of another perforation.
Your patient urgently needs IMMEDIATE treatment!
Reach out to one of the physicians below for help!
Dr. Patrick Whelan
Dr. Beth Kessler
Dr. Lee Shapiro (Albany, NY)
Professor Christos Zouboulis (Germany)
Email contact@degosdisease.org and let us know you’d like to be connected with one of the physicians above.
-

STEP 3
URGENTLY TREAT YOUR PATIENT
No matter how sick your patient is, surgery is not the conclusion of necessary intervention, it is crucial to take immediate action in regard to initiation of medication!!
Reach out to one of the physicians below for help!
Dr. Patrick Whelan
Dr. Beth Kessler
Dr. Lee Shapiro (Albany, NY)
Professor Christos Zouboulis (Germany)
Email contact@degosdisease.org and let us know you’d like to be connected with one of the physicians above.
If the patient is presenting with cutaneous Degos disease and has neurological symptoms, please reach out to Dr. (name), as the treatment differs from that of gastrointestinal presentation.
Differential diagnosis of Degos disease: Why a skin biopsy is needed
Differential diagnosis of Degos disease underscores the necessity of a skin biopsy. This diagnostic step is crucial, particularly in cases where clinical context and morphological features bear a close resemblance.
One notable condition is Fibrosing Dermatomyositis, characterized by guttate-like Gottron’s sign or papules. There have been instances where early papular Degos disease was misdiagnosed as tumid lupus erythematosus, highlighting the importance of thorough and accurate diagnosis.
Evaluating for systemic disease is paramount, as Degos can rapidly evolve from being untreatable to treatable. Without timely intervention, Degos disease can progress swiftly. While treatments for Degos are not FDA-approved, the process of getting them approved can be arduous, often requiring detailed letters to insurance companies. Refer your patients to NIH.
Help your patient pursue referrals to NIH
Why should you preform a laparoscopy?
It is critical a laparoscopy is performed if experiencing abdominal pain. Refer to Gastrointestinal Kohlmeier–Degos disease: a narrative review for images of the lesions.
Laparoscopy Training Video
Patient prognosis
All of the Degos disease literature 15 years ago revealed no positive outcomes, but this has changed with recent treatment advances, and we have the opportunity to make incredible changes in patient outcomes. However, speed is off the essence in terms of diagnosing Degos. Receiving treatment in a timely manner is critical to halt disease progression and spare disease morbidity. If disease stabilization or control can be achieved early, the outcomes will be better. The challenge is getting access to needed medications. It is easier to get the medication if the patient is in the ICU as different approval from insurance companies is needed. Refer to our Treatments & Care page to learn more about drug access.

Support Network
Interested in reaching out to other physicians, check out our Physician Support Network.

Treatments & Care
We have the top experts dedicated to finding treatments for Degos Disease.

Resources
Looking for more information on this ultra-rare disease? Check out or resources section.
Reference
Vazquez- Doval F.J., F.R. De Erenchun, J.A. Paramo, E. Quintanilla (1993). Malignant atrophic papulosis. A report of two cases with altered fibrinolysis and platelet function. Clin. Exp. Dermatol. 18, 441-444